Resultados finales del estudio ACTG 5202: Truvada® o Kivexa® junto a atazanavir/r o efavirenz para el inicio del tratamiento
Xavier Franquet
El miércoles en sesión plenaria, y como presentación de última hora (late breaker), se dieron a conocer los resultados finales del estudio ACTG 5202, un ensayo prospectivo de reparto aleatorio, que ha comparado el uso de las combinaciones de nucleósidos (y nucleótido) tenofovir/emtricitabina (Truvada®) y abacavir/lamivudina (Kivexa®) junto a efavirenz (Sustiva®, y en Atripla® junto a Truvada®) o bien con atazanavir potenciado con ritonavir (Reyataz®/Norvir®) en pacientes sin experiencia previa en el empleo de antirretrovirales.
Eric Daar fue el encargado de ofrecer los resultados generales en la sesión plenaria. Un tiempo atrás, sin embargo, ya se supo que entre las personas que tenían cargas virales altas al empezar el estudio, la eficacia virológica (carga viral indetectable) se perdía antes entre las que tomaban abacavir/lamivudina que entre aquéllas que recibían tenofovir/emtricitabina. Por ello, y a requerimiento del Comité de Seguimiento de Datos y Seguridad [DSMB, en sus siglas en inglés], el estudio dejó de ser ciego para esta franja de participantes.
El 83% de las personas que tomaron parte en esta investigación eran hombres, el 33% negros y el 23% hispanos, con una mediana de edad de 38 años, con una carga viral de 4,7log10 copias/mL y un recuento de CD4 de 230 células/mm3. El seguimiento duró 138 semanas.
Los datos que se han dado a conocer ahora corresponden a la comparación de ambas parejas de nucleósidos (y núcleotido) entre aquellos participantes que empezaron con cargas virales bajas, así como a la comparación de atazanavir/r con efavirenz.
En cuanto a eficacia, en la primera comparación, no se dieron diferencias entre una y otra combinación tanto si se tomaba con efavirenz como con atazanavir/r. Además de estos datos, en la siguiente tabla se pueden ver los resultados de seguridad y tolerabilidad de los participantes que empezaron con cargas virales bajas, así como del total de pacientes.
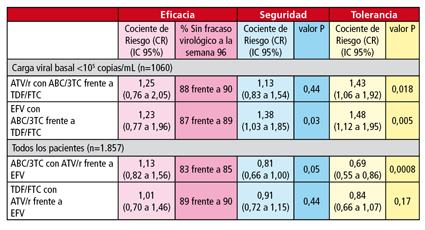
En general, las combinaciones que incluían efavirenz causaron más aumentos de colesterol total, LDL y HDL. En el caso de atazanavir/r, se observó una mayor disminución de la aclaración de la creatinina, cuando se administraba junto a tenofovir, lo que quizá se deba a la interacción entre los dos fármacos, cuyo resultado es el incremento de las concentraciones de ambos.
Según fuentes de la compañía propietaria de atazanavir, Bristol-Myers Squibb, ésta estaría estudiando con detalle las consecuencias de esta alteración en la creatinina; un aspecto importante, teniendo en cuenta que, además, existen planes para una posible coformulación de atazanavir con Truvada® en un solo comprimido.
Por otro lado, ayer, en rueda de prensa, Grace McComsey dio cuenta de los resultados de un subestudio, también presentado en esta conferencia, que midió los cambios en la densidad mineral ósea [DMO] de la cadera y de la columna lumbar y la presencia de lipoatrofia (definida como pérdida de grasa igual o superior al 10% desde el inicio del estudio) en un grupo de 296 participantes, que fueron seguidos durante 96 semanas.
Según McComsey, en los cuatro brazos del estudio se observó una pérdida de DMO, que fue peor durante el primer año, para estabilizarse después. Los brazos con la combinación de tenofovir/emtricitabina fueron los que registraron una mayor pérdida de densidad mineral ósea tanto en la columna lumbar como en el hueso de la cadera.

Las combinaciones con atazanavir/r se asociaron a una mayor pérdida en la columna lumbar en comparación con efavirenz, pero esa diferencia no se observó, sin embargo, en el hueso de la cadera.
En cuanto a los cambios en la grasa corporal, en todos los brazos se dieron incrementos de grasa subcutánea en las extremidades a la semana 96, también en todos se produjeron incrementos de grasa en el tronco, lo que, según McComsey, se interpretó como una vuelta a una cierta normalidad en la distribución de grasas.
Se vieron algunos pocos casos de lipoatrofia en extremidades, con reducciones del 10-20% en el nivel de grasa (5%), que se estabilizaron y no fueron clínicamente significativas. McComsey dijo que se trataba de una gran noticia pues, como aseguró, para que la lipoatrofia sea perceptible por la propia persona deben darse pérdidas de grasa subcutánea superiores, del orden del 25 al 50%.
En declaraciones a La Noticia del Día, la ponente aseguró que no se dio ningún caso de lipoatrofia facial que fuera considerado importante. En cualquier caso, señaló que este subestudio proporcionará más datos próximamente.
Fuente: Elaboración propia.
Referencia: Daar E, Tierney C, Fischl M, et al. ACTG 5202: Final Results of ABC/3TC or TDF/FTC with either EFV or ATV/r in Treatment-naive HIV-infected Patients. 17th Conference on Retroviruses and Opportunistic Infections. February 16-19, 2010. San Francisco. Abstract 59 Late Breaker.
Las personas con VIH logran una reducción importante del riesgo cardiovascular al dejar de fumar
Francesc Martínez / Xavier Franquet
Las tasas de consumo de tabaco entre personas con VIH son muy elevadas. Debido a que en la población general se ha observado una disminución sustancial en el riesgo de padecer enfermedades coronarias tras sólo un año de dejar de fumar, un grupo de investigadores del estudio D:A:D (siglas en inglés de Recopilación de datos sobre efectos adversos de los fármacos anti-VIH) decidió realizar un análisis para averiguar las tasas de acontecimientos cardiovasculares entre aquellos participantes del estudio que hubieran dejado de consumir tabaco.
En el estudio se evaluaron los casos de infarto de miocardio, enfermedades coronarias, enfermedades cardiovasculares (ECV) y la mortalidad por cualquier causa.
Las tasas de dichos acontecimientos se calcularon para no fumadores, ex fumadores en el momento de entrar en el D:A:D, fumadores y ex fumadores que dejaron de consumir tabaco durante el estudio. Los resultados se ajustaron en función de edad, sexo, cohorte, época del año, tratamiento antirretroviral, historial familiar de ECV, presencia de diabetes, presión arterial y niveles de lípidos.

Un total de 27.586 participantes en el estudio fueron incluidos en el análisis. Tras ajustar los resultados en función de las variables anteriormente descritas, el cociente de la tasa de incidencia (IRR, en sus siglas en inglés) de ECV en personas que dejaron de fumar durante el estudio -al ser comparadas con los participantes no fumadores- disminuyó desde 2,32 (IRR durante el primer año posterior al cese del consumo de tabaco) hasta 1,49 después de tres años o más de abandonar el hábito tabáquico. Una tendencia parecida se observó en relación con los casos de infarto de miocardio y enfermedades coronarias. Sin embargo, la disminución del riesgo fue menos pronunciada en el caso de la mortalidad por cualquier causa.
En su presentación de ayer, Katy Petoumenos concluyó que, dada la reducción de riesgos observada en el estudio, las intervenciones sanitarias enfocadas a disminuir el tabaquismo en la población con VIH deben ser consideradas como prioritarias.
Además, en la rueda de prensa anterior a la sesión plenaria señaló también que es preciso que se estudien mejor las posibles interacciones entre los fármacos que se usan para dejar de fumar y los antirretrovirales.
Fuente: Elaboración propia.
Referencia: Petoumenos K, Worm S, Reiss P, et al.Rates of Cardiovascular Disease Following Smoking Cessation in Patients with HIV Infection: Results from the D:A:D Study. 17th Conference on Retroviruses and Opportunistic Infections (CROI 2010). San Francisco, USA. February 16-19, 2010. Abstract 124.
Experiencias en el uso de la profilaxis post-exposición (PPE)
Juanse Hernández
La profilaxis post-exposición (PPE) es una estrategia preventiva cuya finalidad es evitar la infección por VIH después de haber tenido lugar una cierta o probable exposición al virus, y que consiste en la toma de tratamiento antirretroviral durante cuatro semanas. Una sesión de presentaciones ha estado dedicada a abordar experiencias en el empleo de PPE en diferentes contextos y a enfatizar las posibles limitaciones de su uso.
El grupo de estudio DATEMPEP, que aglutina a investigadores de seis centros hospitalarios catalanes (Hospital Clínico, Hospital del Mar y Hospital de San Pablo en Barcelona; Hospital Germans Trias i Pujol en Badalona; Hospital Mutua de Terrassa y Hospital Joan XXIII en Tarragona), ha ofrecido los resultados de un ensayo que evaluó la tasa de interrupción de la PPE antes de las 4 semanas de duración del tratamiento comparando los dos regímenes estándar que se utilizan en el contexto de esta profilaxis: lopinavir/ritonavir (Kaletra®) y atazanavir (Reyataz®), ambos en combinación con zidovudina/lamivudina (AZT/3TC, Combivir®).
Un total de 255 personas que acudieron a los servicios de urgencias de los citados hospitales y que cumplían los criterios de inclusión para tomar PPE fueron distribuidas aleatoriamente para recibir 400/100mg de lopinavir/ritonavir dos veces al día (n= 131) ó 400mg de atazanavir (n= 124) dos veces al día, ambos junto con 250/150mg de AZT/3TC dos veces al día. Además de la tasa de interrupción antes de las 4 semanas, los investigadores también quisieron fijar su atención en el porcentaje de pacientes que no acudían a las citas de seguimiento a los 3 y 6 meses, en el número total de efectos secundarios y en la tasa de seroconversiones.
La media de edad de los pacientes que recibieron PPE fue de 31 años y un 71% eran hombres. El intervalo mediano entre la exposición al VIH y la presentación en el servicio de urgencias fue de 20 horas (de 1 a 72). El tipo de exposición al virus fue mayoritariamente sexual, con una tasa de un 91% (61%, entre hombres que practican sexo con hombres). El nivel de riesgo de infección por VIH fue elevado (0,8-3%) en un 65% de las personas incluidas en el estudio. En 48 personas (19%) se pudo identificar el paciente fuente y 43 de estas personas (17%) tenían infección por VIH.
Los datos muestran que la tasa de pérdida de seguimiento fue muy alta: 77 de los 255 pacientes (30%) que empezaron la PPE no acudieron a la primera cita programada y, por consiguiente, fueron excluidos del análisis. Un total de 178 personas se presentaron a la primera visita, una cifra que fue descendiendo en las sucesivas citas programadas: 159 (día 28), 101 (tercer mes) y 61 pacientes (sexto mes), sin que se observaran diferencias entre los dos brazos de tratamiento.
En trece (7%) de los 178 pacientes [7 en el brazo de lopinavir/ritonavir y 6 en el de atazanavir] la PPE se interrumpió como consecuencia de los efectos secundarios (tiempo mediano tomando tratamiento: 7 días). Un total de 97 de los 178 pacientes (54%) comunicaron efectos secundarios: un 49% (n= 50) en el brazo de lopinavir/ritonavir y un 43% (n= 42) en el de atazanavir [p= 0,59]. No se produjeron seroconversiones entre los 101 y 61 pacientes que acudieron a las visitas del tercer y sexto mes, respectivamente.
Según los investigadores, casi la mitad de los pacientes no completaron los 28 días de PPE recomendados, lo que podría comprometer la eficacia del tratamiento. Además, señalan que la tolerancia fue pobre en ambos brazos: la tasa de efectos secundarios fue de un 49% en el brazo de lopinavir/ritonavir y de un 43% en el de atazanavir. No obstante, en su opinión, este dato no parece explicar la alta tasa de interrupción de la PPE, dado que sólo un 7% de los participantes que recibieron lopinavir/ritonavir y un 10% de los que tomaron atazanavir interrumpieron el tratamiento por los efectos secundarios. Entre los que consiguieron completar la profilaxis, el nivel de adhesión al tratamiento fue elevado (92% en el brazo de lopinavir/ritonavir y 91% en el de atazanavir). En sus conclusiones, los autores afirman que sus hallazgos enfatizan las dificultades de implementar de forma correcta la profilaxis post-exposición en la población general.
Con el fin de mejorar la implementación de la PPE y evitar la interrupción precoz del tratamiento y la pérdida de seguimiento de los pacientes, resultaría útil fortalecer los servicios de counselling e información que se ofrecen a los pacientes que reciben esta estrategia preventiva. Una intervención coordinada entre los centros hospitalarios y los servicios de información y apoyo que proporcionan las organizaciones de base comunitaria permitiría, tal vez, aumentar la eficacia de esta intervención y evitar el abandono del tratamiento por parte de estos pacientes.
Un ejemplo de experiencia en la administración de PPE fuera del contexto estrictamente hospitalario ha sido ofrecido por la Unidad Municipal de Atención de Infecciones de Transmisión Sexual (ITS) de San Francisco. Los resultados de su estudio revelan que la tasa de seroconversión al VIH tras la PPE fue baja, así como también la tasa de interrupción del tratamiento. De hecho, aunque la mayoría de pacientes tomaron PPE por primera vez durante el período de estudio, algunos la habían tomado previamente, e incluso en algunos pacientes era la cuarta vez o más que la recibían. Además, como consecuencia de la incidencia elevada de otras ITS entre los pacientes que acudieron a esta unidad en busca de profilaxis post-exposición, el contexto de las clínicas de atención de enfermedades de transmisión sexual resulta apropiado para realizar intervenciones preventivas adaptadas a las diferentes situaciones de vulnerabilidad frente al VIH y otras ITS.
Fuente: Elaboración propia.
Referencias: Díaz-Brito V, León A, Knobel A, et al. An Open Randomized Multicenter Clinical Trial Comparing Zidovudine/Lamivudine (ZDV/3TC) Plus Lopinavir/r (LPV/r) or Plus Atazanavir (ATV) Used as Postexposure Prophylaxis (PEP) for HIV Infection. 17th Conference on Retroviruses and Opportunistic Infections (CROI 2010). San Francisco, USA. February 16-19, 2010. Abstract 956.
Adamson P, Marcus J, Pipkin S, et al. Characteristics of and HIV Seroconversion Among Patients Receiving Non-Occupational HIV Post-Exposure Prophylaxis in a Sexually Transmitted Disease Clinic – San Francisco, 2007 to 2009. 17th Conference on Retroviruses and Opportunistic Infections (CROI 2010). San Francisco, USA. February 16-19, 2010. Abstract 958.
Tratamiento de la hepatitis aguda en HSH
Juanse Hernández
Hace poco, diversos estudios llevados a cabo en varias ciudades de Europa occidental han evidenciado un aumento en la incidencia de infección aguda por el virus de la hepatitis C (VHC), transmitida muy probablemente por vía sexual, sobre todo en hombres con VIH que mantienen relaciones sexuales con otros hombres (HSH). Sin embargo, hasta la fecha, la investigación en este campo es bastante limitada y no hay directrices basadas en la evidencia sobre el tratamiento de la hepatitis C aguda en personas con VIH. Todavía está por dilucidar cuál es la mejor estrategia de tratamiento en estos pacientes: si utilizar la combinación de interferón pegilado y ribavirina o sólo interferón pegilado o, por lo que respecta a la duración de la terapia, si ésta debe durar 24 ó 48 semanas.
Con el fin de arrojar un poco más de luz sobre cuál podría ser la duración del tratamiento de la hepatitis aguda en hombres con VIH, un grupo de investigadores holandeses realizó un estudio retrospectivo en el que incluyó los datos de HSH con VIH con infección aguda por VHC procedentes de dos centros hospitalarios en Ámsterdam (Holanda).
Para el análisis se dispuso de datos de 53 pacientes con una mediana de edad de 41 años, de los cuales 34 tenían el genotipo 1 del VHC, uno el genotipo 2, uno el genotipo 3, 11 el genotipo 4, y 6, uno desconocido. En el momento del diagnóstico del VHC, el recuento mediano de CD4 fue de 450 células/mm3 y un 98% de pacientes presentaban niveles superiores a 35 U/L de la enzima alanino aminotransferasa (ALT). Un total de 48 participantes completaron el tratamiento, de los cuales en 27 (56%) la duración de la terapia fue de 24 semanas y en 21 (44%), de 48 semanas. En un total de 43 pacientes (90%) la carga viral del VHC fue negativa al finalizar el tratamiento y, según un análisis por intención de tratamiento, 27 de ellos (68%) alcanzaron una respuesta virológica sostenida (RVS; resultado negativo de la carga viral del VHC 6 meses después de concluir el tratamiento).
Según los investigadores, ni el momento de inicio del tratamiento ni su duración se asociaron con los resultados obtenidos. En un análisis estratificado, los responsables del estudio pudieron observar que, cuando se empieza el tratamiento en el transcurso de los seis meses tras la infección, un 82% de los pacientes que recibieron 24 semanas de tratamiento y un 75% de los que lo tomaron durante 48 semanas obtuvieron respuesta virológica sostenida (p >0,6). Cuando el tratamiento se inició más tarde, las tasas de pacientes que lograron una RVS fueron de un 57% (en el grupo de 24 semanas) y de un 82% (en el grupo de 48 semanas) [p >0,2].
En sus conclusiones, los autores afirman que en su estudio no se observó un resultado significativamente mejor ya se empleara una duración más corta o más larga del tratamiento, por lo que sus hallazgos sugieren que un tratamiento durante 24 semanas podría ser suficiente para tratar la infección aguda por VHC en personas coinfectadas por VIH, sobre todo cuando el tratamiento se inicia en el transcurso de los seis meses tras la infección.
Fuente: Elaboración propia.
Referencia: Lambers F, van der Berk G, van der Meer J. Treatment Outcome of Acute Hepatitis C Virus Infection in HIV-infected MSM: The Effect of Treatment Lenghth. 17th Conference on Retroviruses and Opportunistic Infections (CROI 2010). San Francisco, USA. February 16-19, 2010. Abstract 641.
Supervivencia de personas con VIH con cirrosis compensada
Juanse Hernández
Un nuevo análisis de 194 personas con VIH con cirrosis compensada (89% con el virus de la hepatitis C [VHC], 10,3% con el virus de la hepatitis B [VHB], 4,6% con hepatitis delta y un 4,1% con enfermedad hepática por otras causas) a las que se realizó un seguimiento entre octubre de 2004 y diciembre de 2008 en el Hospital Carlos III de Madrid (España) revela una tasa de mortalidad del 5,8% por año, un porcentaje más elevado del que se observa en pacientes cirróticos sin VIH (3-4%) o en personas con VIH sin cirrosis (<2%). Según los investigadores de este estudio, entre los factores asociados con este aumento de la mortalidad figuran una edad avanzada, un recuento bajo de CD4 y una carga viral del VIH detectable en plasma.
Los responsables del estudio señalan que sus hallazgos refuerzan la recomendación actual de proporcionar terapia antirretroviral de gran actividad [TARGA] a todas las personas con VIH que tienen cirrosis, independientemente de sus recuentos de CD4. Además, indican que las puntuaciones en la escala MELD, y sobre todo los valores obtenidos en la prueba de elastometría transitoria (FibroScan), predicen con exactitud la mortalidad en esta población de pacientes.
Fuente: Elaboración propia.
Referencia: Tuma P, Jarrin I, del Amo J, et al. Survival of HIV-Infected Patients with Compensated Liver Cirrhosis. 17th Conference on Retroviruses and Opportunistic Infections (CROI 2010). San Francisco, USA. February 16-19, 2010. Abstract 684.
Raltegravir se muestra seguro y eficaz en pacientes coinfectados
Juanse Hernández
Durante esta edición de la CROI, se han dado a conocer los datos sobre la eficacia y seguridad del inhibidor de la proteasa raltegravir (Isentress®) en personas coinfectadas por el VIH y el virus de la hepatitis B (VHB) y/o C (VHC) que participaron en los estudios de fase III STARTMRK –en pacientes sin experiencia en el uso de tratamientos– y BENCHMRK 1 y 2 –en pacientes pretratados–.
Cada uno de estos estudios fue doble ciego y de distribución aleatoria. En el STARTMRK, los pacientes fueron distribuidos de forma aleatoria para recibir ó 400mg de raltegravir dos veces al día ó 600mg de efavirenz una vez al día, ambos en combinación con tenofovir y emtricitabina (Truvada®). En los estudios BENCHMRK 1 y 2 se incluyeron personas con amplia experiencia en el empleo de tratamiento antirretroviral con virus resistente a múltiples fármacos y opciones terapéuticas limitadas. Los participantes se distribuyeron aleatoriamente para recibir 400mg de raltegravir dos veces al día o bien placebo, ambos administrados con una terapia de base optimizada (TBO).
En estos ensayos se permitió que pudieran participar personas con VIH con infección crónica activa por VHB y/o VHC siempre y cuando los resultados de los análisis basales de la función hepática no excedieran cinco veces el límite superior de normalidad.
En total, 743 pacientes recibieron raltegravir y 519 el fármaco comparador en los tres estudios. Un 16% de los participantes pretratados (144 de 699) y un 6% de los naive (34 de 563) estaban coinfectados por VIH y VHB y/o C (VHB= 6%, VHC= 9%, VHB/VHC= 1% en pretratados; VHB= 4%, VHC= 2%, VHB/VHC= 0,2% en naive).
Los resultados de este análisis a 96 semanas ponen de manifiesto que raltegravir se mostró eficaz tanto en coinfectados por VIH y VHB y/o VHC como en monoinfectados sólo por VIH. El inhibidor de la integrasa fue generalmente bien tolerado en ambos grupos de pacientes; sin embargo, se observaron elevaciones de grado 2, 3 y 4 de las enzimas hepáticas con más frecuencia en los pacientes coinfectados que en los monoinfectados, aunque esta diferencia se pudo apreciar tanto en el grupo que recibió raltegravir como en el grupo control (que tomó efavirenz en el STARTMRK y placebo junto con TBO en los ensayos BENCHMRK).
Con todo, los datos apuntan a que raltegravir puede ser una opción terapéutica apropiada también para las personas coinfectadas por VIH y las hepatitis virales.
Fuente: Elaboración propia.
Referencia: Rockstroh J, Teppler H, Zhao J, et al. Hepatic Safety & Efficacy of Raltegravir in Patients Co-infected with HIV and Hepatitis B (HBV) and/or C (HCV) Virus. 17th Conference on Retroviruses and Opportunistic Infections (CROI 2010). San Francisco, USA. February 16-19, 2010. Abstract 662.
Raltegravir y darunavir consiguen buenas concentraciones en plasma seminal, mientras que maraviroc las alcanza a nivel cerebral
Francesc Martínez
La penetración de los antirretrovirales en los diferentes compartimentos del cuerpo -aspecto importante a la hora de evaluar temas de candente actualidad en el campo del VIH, como la transmisión sexual del virus bajo tratamiento antirretroviral o el deterioro cognitivo asociado a la infección por VIH- suele ser un tema del que se dispone de pocos datos, puesto que la aprobación de los fármacos se ha basado, hasta la fecha, en el análisis de los niveles plasmáticos tanto de los medicamentos como de la carga viral.
Con el fin de incrementar la información relativa a este tema, se han presentado, en esta conferencia, una serie de estudios que evalúan las concentraciones de diferentes fármacos en diversos compartimentos del cuerpo.
Uno de los trabajos presentados evaluó las concentraciones de raltegravir (Isentress®) en el tracto genital femenino (compartimento cervicovaginal), importante tanto en la transmisión sexual como en la vertical [de madre a hijo] del VIH.
Las 14 participantes en el ensayo -mayores de 18 años y con carga viral indetectable durante los tres meses previos al inicio del estudio- recibieron raltegravir (400mg, dos veces al día) junto con una terapia de base optimizada [TBO]. La mediana de la duración del tratamiento fue de 292 días. En el momento de la toma de muestras, todas las participantes tenían una carga viral en el fluido cervicovaginal indetectable y una viremia sanguínea indetectable o muy baja.
La concentración en el fluido cervicovaginal de raltegravir se midió en estado estacionario, alcanzando unos valores 2,3 veces superiores a los de su concentración sanguínea, lo que supone, aproximadamente, una concentración 16 veces superior a la concentración inhibitoria del 95% [CI95] del VIH de genotipo salvaje (VIH WT, en sus siglas en inglés), por lo que cabe suponer un buen control virológico por parte de raltegravir en el compartimento cervicovaginal.
En un segundo estudio, llevado a cabo con voluntarios sanos, se evaluó la concentración de raltegravir en el plasma seminal. Los participantes recibieron raltegravir (400mg, dos veces al día) durante 4 días y una única dosis el quinto día, en el que se tomaron muestras de semen [tras 5 días de abstinencia sexual] y de sangre a las 11-12 horas de la toma de raltegravir (en 4 casos) ó a las 2-4 horas (en otros 4 casos).
El coeficiente de concentración plasmática semen-sangre fue de 1,62 en las mediciones 2-4 horas después de la última toma y de 6,45 en las tomas realizadas tras 11-12 horas, lo que sugiere una buena acumulación y persistencia del fármaco en el compartimento genital masculino.
Un tercer estudio analizó la concentración de darunavir (Prezista®) en el plasma seminal de personas con VIH en monoterapia con darunavir potenciado por ritonavir (n= 23) o el mismo tratamiento junto a dos inhibidores de la transcriptasa inversa análogos de nucleósido (ITIN).
Las concentraciones plasmáticas de darunavir total y libre en sangre a las 12 horas de la última toma fueron, respectivamente, de 3.200 y 212 ng/mL. La concentración de darunavir en plasma seminal tras una mediana de 15,9 horas de la última toma fue de 344 ng/mL. Debido a la similitud observada entre la concentración de fármaco libre en sangre y fármaco total en plasma seminal, y a que la concentración en este último era unas 6 veces la concentración efectiva media corregida en función de las proteínas (CE50CP) del VIH WT, los autores consideraron que darunavir ofrece una buena penetración en el compartimento seminal.
En la misma línea, otro estudio calculó las concentraciones de darunavir en el plasma seminal. En el ensayo se midió la concentración en plasma seminal del fármaco tras 1-3 horas de su administración (11 veces superior a la CE50CP VIH WT), 4-6 horas (9 veces superior a la CE50CP VIH WT) y tras 22-24 horas (4 veces superior a la CE50CP VIH WT). Por lo tanto, en concordancia con el estudio anterior, se consideró que darunavir presenta una concentración eficaz en plasma seminal.
El último de los estudios evaluó la concentración de maraviroc (Celsentri®) en el líquido cefalorraquídeo [LCR] y en el plasma seminal de 12 personas bajo una terapia antirretroviral que incluyera este fármaco.
En el líquido cefalorraquídeo, el fármaco alcanzó niveles similares a los de la concentración inhibitoria del 50% (CI50) del VIH WT e incluso superiores. En semen, a pesar de lograr concentraciones varias veces superiores a la CI50 VIH WT, se observó cierta replicación viral en personas con viremia indetectable en plasma sanguíneo. Por este motivo, serán necesarios nuevos estudios que caractericen la actividad de maraviroc en el plasma seminal.
Fuente: Elaboración propia.
Referencias: Clavel C, Mandelbrot L, Marcelin AG, et al. Raltegravir Concentrations in the Cervicovaginal Compartment Exceed the Median Inhibitory Concentration in HIV-1-infected Women Treated with a Raltegravir-containing Regimen: DIVA 01 Study. 17th Conference on Retroviruses and Opportunistic Infections (CROI 2010). San Francisco, USA. February 16-19, 2010. Abstract 608.
Bonora S, D’Avolio A, Similene M, et al. Steady-state Raltegravir Penetration in Seminal Plasma of Healthy Volunteers. 17th Conference on Retroviruses and Opportunistic Infections (CROI 2010). San Francisco, USA. February 16-19, 2010. Abstract 609.
Lambert-Niclot S, Duvivier C, Algarte-Genin M, et al. Darunavir Concentrations in Seminal Plasma in Patients Receiving Darunavir/ritonavir (DRV/r) Monotherapy: A MONOI-ANRS 136 Substudy. 17th Conference on Retroviruses and Opportunistic Infections (CROI 2010). San Francisco, USA. February 16-19, 2010. Abstract 610.
Taylor S, Jayasuriya A, Dufty N, Guilleran G, et al. Darunavir (DRV) Concentrations Exceed the Protein-corrected (PC) EC50 for Wild Type HIV in the Semen of HIV-1 Positive Infected Men. 17th Conference on Retroviruses and Opportunistic Infections (CROI 2010). San Francisco, USA. February 16-19, 2010. Abstract 611.
Tiraboschi JM, Curto J, Niubo J, et al.Maraviroc Levels in Cerebrospinal Fluid (CSF) and Seminal Plasma from HIV-Infected Patients. 17th Conference on Retroviruses and Opportunistic Infections (CROI 2010). San Francisco, USA. February 16-19, 2010. Abstract 612.




Suscríbete a nuestros boletines
Utiliza este formulario para suscribirte a nuestros boletines. Si tienes cualquier problema ponte en contacto con nosotros.
Al continuar, confirmas que has leído el aviso legal y aceptas la política de privacidad.


